Трисомия 21
Содержание:
Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух
Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала 21-й хромосомы, либо целой хромосомы (трисомия), либо её участков (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от количества дополнительного генетического материала, генетического окружения и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например, был обнаружен у обезьян и мышей). В 2005 году британские исследователи получили анеуплоидных трансгенных мышей с наличием 21-й человеческой хромосомы в дополнение к стандартному набору мышей. Нормальный человеческий кариотип содержит 46 хромосом и обозначается 46,XY у мужчин и 46,XX у женщин, в то время как у носителей синдрома Дауна с трисомией по 21-й хромосоме кариотип содержит 47 хромосом.
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме.
Риск рождения ребёнка с синдромом Дауна и другими численными хромосомными аномалиями растёт с возрастом матери. Точная причина этого неизвестна, но, по-видимому, она связана с возрастом яйцеклеток матери.
Трисомия происходит из-за нерасхождения хромосом во время мейоза, в результате чего возникает гамета с 24 хромосомами. При слиянии с нормальной гаметой противоположного пола образуется зигота с 47 хромосомами, а не 46-ю, как без трисомии.
Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских.
Мозаицизм
Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является, как правило, более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики.
По данному типу синдром появляется в 1—2 % случаев.
Робертсоновские транслокации
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться вследствие наличия робертсоновской транслокации в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й ). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна. Эта форма не зависит от возраста матери. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Дупликация части хромосомы 21
Очень редко участки 21-й хромосомы могут быть удвоены в результате хромосомной перестройки. При этом возникают дополнительные копии некоторых, но не всех генов из 21-й хромосомы. Если продублируются фрагменты, обусловливающие физические и психологические проявления синдрома Дауна, то ребёнок родится с этим синдромом. Такие хромосомные перестройки происходят крайне редко, и не существует оценки периодичности данного явления.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант синдрома — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % людей с синдромом Дауна наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.
Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет.
Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала по 21-й хромосоме, либо полностью (трисомия), либо частично (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от степени копии, генетической истории и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например был обнаружен у обезьян и мышей). Совсем недавно[когда?
] исследователи[кто? ] вывели трансгенных мышей с наличием 21-й человеческой хромосомы (в дополнение к стандартному набору мышей). Добавление генетического материала может проводиться в разных направлениях. Типичный человеческий кариотип обозначается как 46,XY (мужской) или 46,XX (женский) (различие в поле несёт Y-хромосома).
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме.
Синдром Дауна и сходные хромосомные аномалии чаще встречаются у детей, рождённых немолодыми женщинами. Точная причина этого неизвестна, но, по-видимому, она как-то связана с возрастом яйцеклеток матери.
Трисомия происходит из-за того, что во время мейоза хромосомы не расходятся. При слиянии с гаметой противоположного пола у эмбриона образуется 47 хромосом, а не 46, как без трисомии.
Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских.
Мозаицизм
Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является как правило более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики.
По данному типу синдром появляется в 1—2 % случаев.
Робертсоновские транслокации
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться за счёт робертсоновских транслокаций в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й ). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции, нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна
. Это не зависит от возраста матери и показывает скорее равную роль родительских организмов в появлении синдрома Дауна. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Дублирование части хромосомы 21
Очень редко, но части хромосом могут делиться. Это создаст дополнительные копии некоторых, но не всех генов из 21-й хромосомы. Если продублируются фрагменты, обусловливающие физические и психологические проявления синдрома Дауна, то ребёнок родится с этим синдромом. Такое дублирование происходит крайне редко и не существует оценки периодичности данного явления.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.
Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет.
Уровни компактизации хромосомной ДНК
См. также: Сверхспирализация ДНК
Основу хромосомы составляет линейная макромолекула ДНК значительной длины. В молекулах ДНК хромосом человека насчитывается от 50 до 245 миллионов пар азотистых оснований. Суммарная длина ДНК из одной клетки человека составляет величину порядка двух метров. При этом типичное ядро клетки человека, которое можно увидеть только при помощи микроскопа, занимает объём около 110 мкм³, а митотическая хромосома человека в среднем не превышает 5—6 мкм. Подобная компактизация генетического материала возможна благодаря наличию у эукариот высокоорганизованной системы укладки молекул ДНК как в интерфазном ядре, так и в митотической хромосоме. Надо отметить, что у эукариот в пролиферирующих клетках осуществляется постоянное закономерное изменение степени компактизации хромосом. Перед митозом хромосомная ДНК компактизуется в 105 раз по сравнению с линейной длиной ДНК, что необходимо для успешной сегрегации хромосом в дочерние клетки, в то время как в интерфазном ядре для успешного протекания процессов транскрипции и репликации хромосоме необходимо декомпактизоваться. При этом ДНК в ядре никогда не бывает полностью вытянутой и всегда в той или иной степени упакована. Так, расчётное уменьшение размера между хромосомой в интерфазе и хромосомой в митозе составляет всего примерно 2 раза у дрожжей и 4—50 раз у человека.
Упаковка ДНК в хроматин обеспечивает многократное сокращение линейных размеров ДНК, необходимое для размещения её в ядре. Она происходит в несколько этапов. Наиболее изученными являются три первых уровня упаковки: (1) накручивание ДНК на нуклеосомы с образованием нуклеосомной нити диаметром 10 нм, (2) компактизация нуклеосомной нити с образованием так называемой 30-нм фибриллы и (3) сворачивание последней в гигантские (50 — 200 тысяч п. н.) петли, закреплённые на белковой скелетной структуре ядра — ядерном матриксе.
Одним из самых последних уровней упаковки в митотическую хромосому некоторые исследователи считают уровень так называемой хромонемы, толщина которой составляет около 0,1—0,3 мкм. В результате дальнейшей компактизации диаметр хроматиды достигает ко времени метафазы 700 нм. Значительная толщина хромосомы (диаметр 1400 нм) на стадии метафазы позволяет, наконец, увидеть её в световой микроскоп. Конденсированная хромосома имеет вид буквы X (часто с неравными плечами), поскольку две хроматиды, возникшие в результате репликации, соединены между собой в районе центромеры (подробнее о судьбе хромосом при клеточном делении см. статьи митоз и мейоз).
Триплоидии
Крайне редко наблюдаемые при мертворождениях, триплоидии составляют пятую по частоте хромосомную аномалию в материале выкидыше. В зависимости от соотношения половых хромосом может быть 3 варианта триплоидий: 69XYY (самая редкая), 69, XXX и 69, XXY (самая частая). Анализ полового хроматина показывает, что при конфигурации 69, XXX чаще всего обнаруживается только одна глыбка хроматина, а при конфигурации 69, XXY чаще всего половой хроматин не обнаруживается.
Приведенный ниже рисунок иллюстрирует различные механизмы, приводящие к развитию триплоидии (диандрию, дигинию, диспермию). С помощью специальных методов (хромосомные маркеры, антигены тканевой совместимости) удалось установить относительную роль каждого из этих механизмов в развитии триплоидии у зародыша. Оказалось, что на 50 случаев наблюдений триплоидия была следствием дигинии в 11 случаях (22%), диандрии либо диспермии — в 20 случаях (40%), диспермии — в 18 случаях (36%).
| Механизмы образования триплоидной зиготы |
|---|
Двойные аберрации
Большая частота хромосомных аномалий в материале выкидышей объясняет высокую частоту комбинированных аномалий в одном и том же зародыше. Напротив, у новорожденных комбинированные аномалии крайне редки. Обычно в таких случаях наблюдаются комбинации аномалии половой хромосомы и аномалии аутосомы.
В связи с более высокой частотой аутосомных трисомий в материале выкидышей, при комбинированных хромосомных аномалиях у абортусов чаще всего встречаются двойные аутосомные трисомии. Трудно сказать, связаны ли такие трисомии с двойным «нон-дисджанкшн» в одной и той же гамете, или со встречей двух аномальных гамет.
Частота сочетаний различных трисомий в одной и той же зиготе носит случайный характер, что позволяет предположить независимость друг от друга появления двойных трисомий.
Комбинация двух механизмов, приводящих к появлению двойных аномалий, позволяет объяснить появление других аномалий кариотипа, встречающихся при выкидышах. «Нон-дисджанкшн» при образовании одной из гамет в сочетании с механизмами образования полиплоидии объясняет появление зигот с 68 или 70 хромосомами. Сбой первого митотического деления у такой зиготы с трисомией может приводить к таким кариотипам, как 94,XXXX,16+,16+.
Пренатальная диагностика
1) Инвазивные исследования (амниоцентез, биопсия хориона) в основном назначают тем женщинам, у которых наблюдается повышенный риск того, что родится малыша с синдромом Эдвардса, например, пациенткам, чей возраст превышает 35 лет или с плохими результатами неинвазивных тестов: УЗИ и анализов. Инвазивные методы диагностики являются высокоточными, однако, учитывая риск осложнений, не подходят для массового проведения всем беременным, а проводятся только по особым показаниям.
2) Неинвазивные технологии, так называемые скрининги. Скрининг – комплексное исследование беременных женщин на наличие у плода хромосомных аномалий. Выделено несколько признаков, указывающих на высокий риск наличия заболевания, которые может выявить УЗИ плода (отсутствие носовой кости, увеличенная толщина воротникового пространства, недостаточная длина бедренных и плечевых костей и другие особенности). В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A. Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
Однако при использовании стандартных тестов на синдром Эдвардса, лишь у 3% женщин, направленных на инвазивную диагностику действительно подтверждается наличие заболевания. В то же время не исключены и ложно-отрицательные результаты, когда скрининг показывает низкий риск, а ребенок рождается с хромосомной патологией.
- точность 99%, что намного точнее классической диагностики (УЗИ и биохимический скрининг)
- совершенно безопасен, в отличие от инвазивных методик — для забора материала на анализ необходимо просто взять кровь из вены беременной женщины.
- на ранних сроках: анализ можно проводить уже на 9-й неделе беременности.
Показания для тестирования на ломкую Х-хромосому
- Индивиды с задержкой умственного и общего развития, аутизмом
- Индивиды с чертами фрагильной Х-хромосомы
- Индивиды с наличием синдрома фрагильной Х-хромосомы в семейном анамнезе
- Индивиды с наличием в семейном анамнезе недиагностированной задержки умственного развития
- Плоды от матерей-носителей
Геномный импринтинг — процесс, при котором активация гена происходит преимущественно в материнской или преимущественно в родительской хромосоме, но не в обеих хромосомах. Нормальное развитие имеет место лишь в том случае, если присутствуют обе копии (материнская и отцовская) импринтинг-ген. Импринтинг-ген неактивен, значит, активный ген теряет (путем делеции) или получает мутацию, в таком случае плод будет пораженным. Лишь несколько генов могут испытывать импринтинга. Примерами геномного импринтинга может быть синдром Ангельмана и полный пузырный занос (вариант гестационной трофобластической болезни).
Синдром Ангельмана характеризуется тяжелой задержкой умственного развития, атаксической походкой, типичным лицом, пароксизмами смеха и судорогами. Ген синдрома Ангельмана является активным только в материнской унаследованной хромосоме, следовательно, если происходит делеция материнской хромосомы 15 или материнская копия гена имеет мутацию, белковый продукт не образуется и плод будет пораженным.
Синдром Ангельмана также может развиться, если обе копии хромосомы 15 является унаследованными от отца (отсутствие материнской копии хромосомы 15). Это состояние получило название унипарентальной дисомии. Унипарентальная дисомия возникает чаще вследствие потери хромосомы у эмбриона с трисомией или добавления хромосомы у плода с моносомией по этой хромосомой. Каждая из хромосом может быть генетически различной (гетеродисомия) или идентичной (изодисомия), в зависимости от времени возникновения этого феномена — в течение первого или второго мейотического деления, соответственно.
Полный пузырный занос обычно является диплоидным (46, ХХ или Х ¥), но может иметь полностью отцовское происхождение, без материнского хромосомного материала. При таких условиях плод не может развиваться. Полный пузырный занос может сопровождать нормальную многоплодную беременность, но в этом случае возрастает риск материнских осложнений (гипертиреоидизм, преэклампсия, преждевременные роды). В отличие от полного пузырного заноса, частичный пузырный занос обычно является триплоидным (69, ХХХ, 69, ХVV), с дополнительным набором отцовских хромосом.
Триплоидия с дополнительным набором материнских хромосом имеет место при ЗВУР плода, врожденных пороках развития и маленькой плаценте.
Факторы, напрямую связанные с родителями
Влияние возраста матери на вероятность рождения ребенка с трисомией 21 наводит на мысль о возможном влиянии возраста матери на вероятность возникновения летальных хромосомных аберраций у зародыша. Приводимая ниже таблица показывает связь возраста матери с кариотипом материала выкидышей.
| Средний возраст матери при хромосомных аберрациях абортусов | ||
|---|---|---|
| Кариотип | Число наблюдений | Средний возраст |
| Нормальный | 509 | 27,5 |
| Моносомия X | 134 | 27,6 |
| Триплоидии | 167 | 27,4 |
| Тетраплоидия | 53 | 26,8 |
| Аутосомные трисомии | 448 | 31,3 |
| Трисомии D | 92 | 32,5 |
| Трисомии E | 157 | 29,6 |
| Трисомии G | 78 | 33,2 |
Как видно из таблицы, не было обнаружено связи между возрастом матери и самопроизвольными выкидышами, связанными с моносомией X, триплоидией или тетраплоидией. Повышение среднего возраста матери отмечено для аутосомных трисомий в целом, но по разным группам хромосом цифры были получены разные. Однако общее число наблюдений в группах недостаточно, чтобы уверенно судить о каких-либо закономерностях.
Возраст матери в большей степени связан с повышенным риском выкидышей с трисомиями акроцентрических хромосом группы D (13, 14, 15) и G (21, 22), что совпадает и со статистикой хромосомных аберраций при мертворождениях.
Для некоторых случаев трисомий (16, 21) было определено происхождение лишней хромосомы. Оказалось, что возраст матери связан с повышением риска трисомий только в случае материнского происхождения лишней хромосомы. Не было обнаружено связи возраста отца с повышением риска трисомий.
В свете исследований на животных высказываются предположения о возможной связи старения гамет и задержки оплодотворения на риск возникновения хромосомных аберраций. Под старением гамет понимают старение сперматозоидов в половых путях женщины, старение яйцеклетки либо в результате перезрелости внутри фолликула или в результате задержки выхода яйцеклетки из фолликула, либо в результате трубной перезрелости (запоздалого оплодотворения в трубе). Скорее всего, подобные законы действуют и у человека, но достоверных подтверждений этого пока не получено.
Оценка частоты хромосомных нарушений в момент зачатия
Можно попробовать расчитать количество зигот с хромосомными аномалиями при зачатии, основываясь на частоте хромосомных аномалий, обнаруживаемых в материале выкидышей. Однако прежде всего следует отметить, что поразительное сходство результатов исследований материала выкидышей, проведенное в разных частях света, говорит о том, что хромосомные сбои в момент зачатия являются очень характерным явлением в репродукции у человека. Кроме того, можно констатировать, что реже всего встречающиеся аномалии (например, трисомии A, B и F) связаны с остановкой развития на очень ранних стадиях.
Анализ относительной частоты различных аномалий, возникающих при нерасхождении хромосом в процессе мейоза, позволяет сделать следующие важные выводы:
1. Единственной моносомией, обнаруживаемой в материале выкидышей, является моносомия X (15% всех аберраций). Напротив, аутосомные моносомии практически не обнаруживаются в материале выкидышей, хотя теоретически их должно быть столько же, сколько и аутосомных трисомий.
2. В группе аутосомных трисомий частота трисомий разных хромосом значительно варьирует. Исследования, выполненные с использованием метода G-бэндинга, позволили установить, что все хромосомы могут быть участницами трисомии, однако некоторые трисомии встречаются гораздо чаще, например, трисомия 16 встречается в 15% случаев всех трисомий.
Из этих наблюдений можно сделать вывод, что, скорее всего, частота нерасхождения разных хромосом приблизительно одинакова, а различная частота аномалий в материале выкидышей связана с тем, что отдельные хромосомные аберрации приводят к остановке развития на очень ранних стадиях и поэтому с трудом поддаются обнаружению.
Эти соображения позволяют приблизительно расчитать реальную частоту хромосомных нарушений в момент зачатия. Расчеты, сделанные Буэ, показали, что каждое второе зачатие дает зиготу с хромосомными аберрациями.
Данные цифры отражают среднюю частоту хромосомных аберраций при зачатии в популяции. Однако данные цифры могут значительно колебаться у разных супружеских пар. У некоторых супружеских пар вероятность возникновения хромосомных аберраций в момент зачатия значительно превышает средний риск в популяции. У таких супружеских пар невынашивание беременности на малых сроках происходит гораздо чаще, чем у остальных супружеских пар.
Данные расчеты подтверждаются другими исследованиями, проведенными с использованием других методов:
1. Классическими исследованиями Хертига
2. Определением уровня хорионического гормона (ХГ) в крови женщин после 10 после зачатия. Часто этот тест оказывается положительным, хотя менструация приходит вовремя или с небольшой задержкой, и субъективно наступления беременности женщина не замечает («биохимическая беременность»)
3. Хромосомный анализ материала, полученного при искусственных абортах показал, что при абортах на сроке 6—9 недель (4—7 недель после зачатия) частота хромосомных аберраций составляет примерно 8%, а при искусственных абортах на сроке 5 недель (3 недели после зачатия) эта частота возрастает до 25%.
4. Было показано, что нерасхождение хромосом в процессе сперматогенеза является очень частым явлением. Так Пирсон и сотр. обнаружили, что вероятность нерасхождения в процессе сперматогенеза для 1-й хромосомы составляет 3,5%, для 9-й хромосомы — 5%, для Y-хромосомы — 2%. Если и другие хромосомы имеют вероятность нерасхождения примерно такого же порядка, то тогда только 40% всех сперматозоидов имеют нормальный хромосомный набор.
Влияние хромосомных аномалий на развитие зиготы
Хромосомные аномалии зиготы проявляются как правило уже в первые недели развития. Выяснение конкретных проявлений каждой аномалии сопряжено с целым рядом трудностей.
Во многих случаях установление срока беременности при анализе материала выкидышей крайне затруднено. Обычно сроком зачатия считается 14-й день цикла, но у женщин с невынашиванием беременности часто бывают задержки цикла. Кроме того, очень трудно бывает установить дату «смерти» плодного яйца, поскольку от момента гибели до выкидыша может пройти много времени. В случыае триплоидии этот период может составить 10—15 недель. Применение гормональных препаратов может еще более удлиннить это время.
С учетом этих оговорок, можно сказать, что чем меньше срок беременности на момент гибели плодного яйца, тем выше частота хромосомных аберраций. Согласно исследованиям Кризи и Лоритсена, при выкидышах до 15 недель беременности частота хромосомных аберраций составляет около 50%, при сроке 18 — 21 неделя — около 15%, при сроке более 21 недели — около 5—8%, что примерно соответствует частоте хромосомных аберраций в исследованиях перинатальной смертности.
Признаки Синдрома Патау
- низкая масса тела при доношенной беременности (менее 2500 грамм);
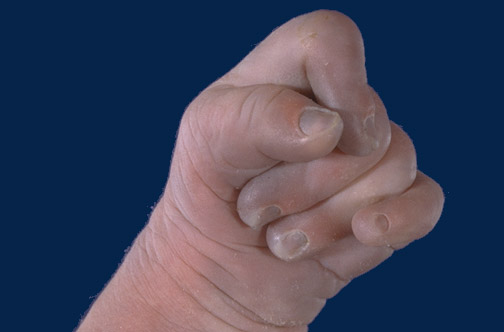
- неправильное строение черепа (небольшие размеры, сужение лба, расширение затылочной области);Выраженные отклонения физического и умственного развития;
- пороки развития структур головного мозга;
- пороки развития глаз (отсутствие глазных яблок, маленькие глаза, катаракта, отслойка сетчатки и др.);
- заячья губа;
- волчья пасть;
- деформированная форма ушей;
- пороки развития кисти (лишние пальцы, неправильное формирование большого пальца);
- локальное отсутствие кожи, волос;
- деформированные стопы, лишние пальцы на ногах;
- множественные пороки мочевыводящей, сердечно-сосудистой, пищеварительной и половой систем.
Инвазивные методы определения трисомии по 21-й хромосоме
Хорионическая биопсия – лабораторный метод, который предполагает забор ворсинок и клеток плаценты с помощью катетера.
Забор пуповинной крови.
Инвазивные методы позволяют полностью исключить наличие заболевания, однако, всегда существует вероятность риска прерывания беременности.
Помимо инвазивных и неинвазивных методов существует еще один способ диагностики трисомии. Им является пренатальный ДНК тест, который можно провести уже в первом триместре. Метод является полностью безопасным и на 100% информативным. Подробная расшифровка молекулы ДНК занимает 14 дней. После этого родителям озвучивают результат.