Фенилкетонурия — симптомы и лечение
Содержание:
Окончание диетотерапии
Ранее считалось, что дети могут прекратить диету по достижении 5-6 летнего возраста.
Последние исследования показали, что дети с фенилкетонурией, прекратившие лечение в детстве, не могут развиваться также, как дети, продолжающие диетотерапию, и имеют нарушения в обучении, поведении, другие неврологические проблемы.
В настоящее время многие авторы склонны считать необходимым поддерживать ограничительную диету при ФКУ пожизненно.
До тех пор, пока не будет предложено другое лечение, все больные с ФКУ должны придерживаться ограничительной диеты для поддержания безопасного уровня фенилаланина (2-6 мг%). (Schuett, 1996).
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Online-консультации врачей
| Консультация трихолога (лечение волос и кожи головы) |
| Консультация специалиста банка пуповинной крови |
| Консультация пластического хирурга |
| Консультация детского психолога |
| Консультация аллерголога |
| Консультация педиатра |
| Консультация ортопеда-травматолога |
| Консультация эндоскописта |
| Консультация пульмонолога |
| Консультация андролога-уролога |
| Консультация сексолога |
| Консультация специалиста по лечению за рубежом |
| Консультация радиолога (диагностика МРТ, КТ) |
| Консультация диетолога-нутрициониста |
| Консультация анестезиолога |
Новости медицины
6 простых привычек, чтобы круглый год не болеть простудами: рекомендуют все врачи,
17.03.2021
Морепродукты становятся вредными для здоровья?,
05.01.2021
Digital Pharma Day. Будьте во главе digital-трансформации фармацевтической индустрии,
09.10.2020
В сети EpiLaser самые низкие цены на ЭЛОС эпиляцию в Киеве,
14.09.2020
Новости здравоохранения
Эксперт назвала три отличия простуды от COVID-19,
05.01.2021
В мире более 86 миллионов случаев COVID-19,
05.01.2021
Скорость распространения COVID-19 зависит от климатических условий,
11.06.2020
Исследователи насчитали шесть разновидностей коронавируса,
11.06.2020
Причины боли в тазобедренном суставе
Как отмечают врачи, боли в таком подвижном соединении достаточно сложны в диагностике. Часто симптомы наблюдаются как у взрослых, так и у детей. Существует несколько основных причин такого состояния:
- Травмы. Ушибы, разрывы, растяжения, переломы, трещины. В этом случае пациент сам часто может установить источник недуга, так как прислушивается к своим ощущениям.
- Болезни. Синдром грушевидной мышцы, бурсит, хондроматоз, теносиновит, остеопороз, остеонекроз и многие другие. Болезни поражают как сам сустав, так и связки, окружающие ткани, мышцы. Вне зависимости от области поражения, больной будет чувствовать неприятные ощущения именно в тазобедренной области. Выявление причины – более сложная задача, требующая применения современных методов диагностики.
- Различные причины иррадирующей боли. Нередко боли в тазобедренном суставе возникают и при заболевании других органов (например, позвоночника или области паха).
- Заболевания, носящие системный характер. Фибромиалгия, лейкемия, спондилоартрит и многие другие. В этом случае также разрабатывается системный подход к лечению пациента.
Стоит особо отметить тот факт, что ранняя диагностика причин заболевания тазобедренного сустава – это большой шаг к организации правильного и своевременного лечения.
Фенилкетонурия: механизм заболевания
Один из ингредиентов нашей еды — белки. Белки, как растительные (например, в картофеле, рисе, маргарине), так и животные (мясо, рыба, яйца), состоят из мелких частиц, похожих на кирпичи в стене. Затем во время пищеварения эти белки расщепляются на различные строительные блоки, из которых тело строит необходимые структуры, например, мышцы или гормоны.
У пациентов с фенилкетонурией отсутствует фермент, ответственный за преобразование одного из этих строительных блоков: фенилаланина. В результате этого нарушения фенилаланин не может быть превращен в другой строительный блок, тирозин.
В результате фенилаланин и продукты его аномального метаболизма начинают накапливаться в крови ребенка, страдающего фенилкетонурией. В организме тирозин необходим для образования гормонов щитовидной железы (тироксин), гормонов надпочечников (адреналин, норадреналин), для производства красителей (меланинов — это пигмент кожи человека) и для построения других белков организма. У ребенка с фенилкетонурией уровень фенилаланина в крови повышается, а количество тирозина недостаточно для правильного функционирования организма.
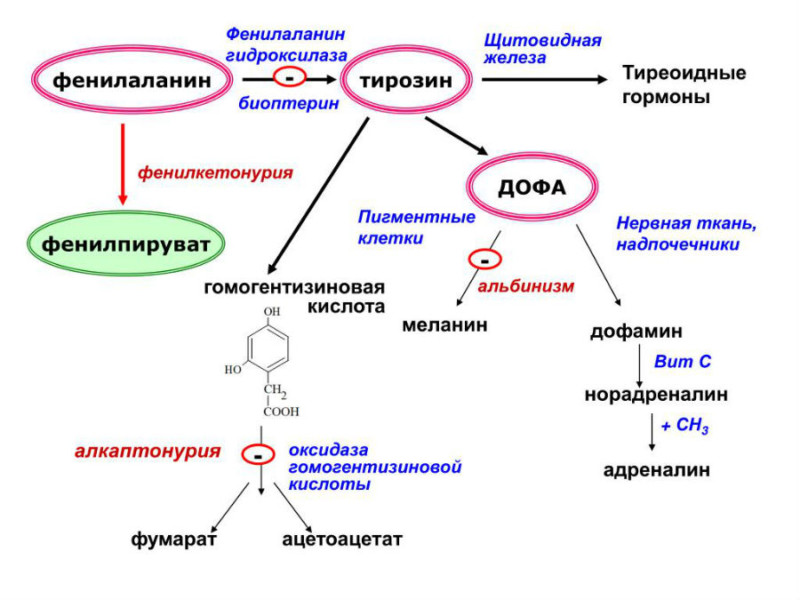 Метаболизм фенилаланина
Метаболизм фенилаланина
Организм находит другой способ избавиться от избытка фенилаланина — вместо тирозина он превращается в такие соединения, как фенилпировиноградная кислота, фенилмолочная кислота и гидроксифенилуксусная кислота. Эти кислоты тоже являются кетонами, отсюда и название болезни — фенилкетонурия.
Они накапливаются в крови, а затем появляются в моче. Почти в 98% случаев дефект связан с мутацией в гене, кодирующем фермент фенилаланингидроксилазу (ПАГ), а в остальных случаях возникают мутации в генах, кодирующих ферменты, связанные с биосинтезом или метаболизмом кофактора реакции превращения фенилаланина в тирозин.
В Европе заболеваемость фенилкетонурией в среднем составляет 1 из 7 тысяч. живорождений. Это генетическое заболевание, переданное ребенку обоими родителями.
Заболевание вызвано мутацией генов на 12-й хромосоме, которые кодируют ферменты в системе гидроксилирования фенилаланина. Выявлено около 800 различных мутаций в генах, кодирующих фенилаланингидроксилазу (PAH), GTP-циклогидролазу I (GTPCH), 6-пирувилтетрагидробиоптеринсинтазу (PTS) и тетрагидробиоптеринредуктазу (DHPR). – мутации этих генов связаны с заболеванием. Самые распространенные из них касаются гена, кодирующего белок PAH.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Диагностика
Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов распада фенилаланина (фенилкетонов) в моче (не ранее 10—12 дня жизни ребёнка). Также возможно определение активности фермента фенилаланингидроксилазы в биоптате печени и поиск мутаций в гене фенилаланингидроксилазы.
Для диагностики 2 и 3 типа, связанных с мутацией в гене, отвечающем за синтез кофактора, необходимы дополнительные диагностические исследования.
В возрасте от 2—4 месяцев у больных появляются такие симптомы, как вялость, судороги, гиперрефлексия, «мышиный» запах пота и мочи или «запах волка», экзема. А также среди других симптомов отмечены: мышечная гипертензия, гиперкинезы, неустойчивая походка, при несоблюдении диеты светлеют глаза, волосы, кожа (по причине недостаточного количества в организме меланина, производного тирозина); судорожные припадки.
Психическое состояние
Фенилкетонурия сопровождается глубокой степенью умственной отсталости, обычно идиотией или имбецильностью. Могут наблюдаться явления эхопраксии (повторение движений окружающих) и эхолалии (повторение речи). Для больных фенилкетонурией характерна вялость с редкими вспышками злобы и раздражительности.
Диагностика фенилкетонурии
Природа – весьма непредсказуемая и удивительная вещь.
Огромному количеству людей она приносит одни лишь радости и дарит им совершенно здоровых детей, других же обделяет, преподнося им своего рода «сюрпризы» жизни. Одним из таких сюрпризов принято считать сферу генетических закономерностей, а точнее заболеваний, которые возникают вследствие наследственных мутаций генов.
Особенно часто дети с различными врожденными патологиями рождаются в весьма благополучных семьях. В таких случаях родители просто не знают о том, что у них в организме имеется так называемый патологический задаток. Одной из таких наследственных патологий является заболевание под название фенилкетонурия.
Каковы же методы диагностики данного недуга?
Ни для кого не секрет, что новорожденный начинает расти и развиваться с самых первых дней жизни. До недавнего времени было принято считать, что данную патологию на самом раннем этапе ее развития выявить невозможно. На сегодняшний же день вопрос своевременного выявления данного заболевания у младенцев принято считать решенным.
Все последние десять лет ученые занимались данным вопросом и наконец-то нашли правильный подход к решению имеющейся проблемы.
Современная медицина предоставляет так называемый скрининг-тест, который представляет собой метод массового обследования младенцев на наличие непосредственно фенилкетонурии. Данный тест является ничем иным как анализом крови. Пройти данный тест можно в каждом центре генетики и в родильных домах.
Уже на третий – пятый день жизни новорожденного у него через три часа после последнего кормления берут из пятки всего лишь несколько капель крови, которую впоследствии наносят на специальный бумажный бланк, имеющийся в каждой лаборатории. Капли крови следует наносить на бумажный бланк таким образом, чтобы получилось всего три кружка, диаметр которых должен быть не меньше двенадцати миллиметров. Очень важен еще и тот факт, чтобы эти кружки были пропитаны кровью полностью.
В последующем этот бланк вместе с кровью отправляют в лабораторию скрининга, специалисты которой проводят ее анализ, посредством которого удается выявить истинное количество фенилаланина в сыворотке крови.
Данный анализ проводят в течение суток, так что Вы сможете ознакомиться с его результатом уже через двадцать четыре часа с момента, как он попадет в лабораторию скрининга. Чем раньше будет взят анализ крови, тем лучше. И еще, проследите за тем, чтобы данные скрининг-теста были записаны в обменную карту ребенка
Это очень важно.
Если же в родильном доме по определенным причинам данный анализ сдать не удалось, тогда запишитесь на консультацию к генетику, который выдаст Вам направление на проведение данного теста.
В случае если результат анализа является положительным, тогда Вас в обязательном порядке попросят сдать его еще раз. Если и повторное тестирование укажет на наличие данной патологии, тогда придется начинать весьма продолжительный курс терапии данного недуга.
Паниковать в таких случаях не нужно.
|
Следуйте всем рекомендациям специалиста и тогда вполне возможно, что уже через несколько лет Вам удастся забыть о фенилкетонурии раз и навсегда. |
К каким докторам следует обращаться если у Вас Фенилкетонурия (ФКУ) у детей:
Педиатр
Психоневролог
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Фенилкетонурии (ФКУ) у детей, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Диагностика
Диагностика лактазной недостаточности осуществляется на основании данных анамнеза, клинического осмотра, сбора жалоб, лабораторных (включая генетические) и инструментальных методов обследования. Диагностика направлена на определение формы заболевания, тяжести состояния и возникающих осложнений.
Из данных анамнеза можно выяснить (при вторичной недостаточности) наличие кишечных инфекций (бактериальных, вирусных (ротавирус), глистные инвазии, наличие простейших (амебиаз), хронические воспалительные заболевания кишечника (болезнь Крона, язвенный колит, целиакия), пищевая аллергия.
Лабораторными методами диагностики являются определение содержания углеводов в кале; нагрузочный тест с лактозой; исследование генов, отвечающих за синтез фермента лактазы в клетках тонкого кишечника; определение содержания водорода, метана или меченого углерода (СО2) в выдыхаемом воздухе (методы отражают активность микрофлоры по расщеплению лактозы).
Нагрузочный тест с лактозой предусматривает определение уровня сахара в крови до и после перорального приема лактозы (в норме в течение 60 минут после приема лактозы содержание глюкозы в крови должен повыситься не менее чем на 20 % от исходного).
Наиболее достоверным тестом в диагностике ЛН является определение активности фермента — лактазы в биоптатах слизистой оболочки тонкой кишки.
Молекулярно-биологическое исследование гена МСМ6 используется для выявления генетической предрасположенности к непереносимости лактозы (ген МСМ6 регулирует ген LCT, который кодирует фермент лактазу, расщепляющей сахар — лактозу). Носители генотипа С/С характеризуются непереносимостью к лактозе, генотип С/Т характеризуется вариабельной активностью лактазы (т.е. существует риск развития недостаточности лактазы). Наличие генотипа Т/Т свидетельствует о хорошей переносимости к лактозе.
Косвенным свидетельством лактазной недостаточности является снижение рН (кислотности) кала (в норме 5,5 и выше).
Клинический анализ крови, биохимическое исследование проводятся для оценки текущего состояния пациента, диагностики и мониторирования сопутствующих или основного заболевания.
Одним и методов подтверждения наличия лактазной недостаточности является перфузионный метод исследования кишечного пищеварения и всасывания.
Дифференциальная диагностика проводится с заболеваниями и состояниями, протекающими с нарушением всасывания питательных веществ в тонком кишечнике. К ним относятся болезнь Крона, болезнь Уиппла, врожденная короткая кишка, врожденная натриевая диарея (редкое заболевание, сопровождающееся поносами и снижением уровня натрия в организме), врожденная недостаточность фермента амилазы, гипертиреоз (гиперфункция щитовидной железы), гипопаратиреоз (недостаточная активность паращитовидных желез), гормон-продуцирующие опухоли (гастринома, нейробластома, карциноид и др), муковисцидоз (хроническое заболевание с нарушением работы желез внешней секреции), целиакия, непереносимость различных белков коровьего молока, пищевая аллергия и многие другие.
Основные используемые лабораторные исследования:
- Определение активности лактазы в биоптатах слизистой оболочки тонкой кишки.
- Содержание углеводов в кале.
- Общий анализ кала (копрограмма с определением рН).
- Генетические исследования: лактазная недостаточность (Adult Lactase Deficiency (Gene MCM6)), анализ полиморфизма c.-13910C>T в гене MCM6 (регуляторная область гена LCT).
- Гистологическое исследование слизистой тонкого кишечника (небольшой участок слизистой изучают под микроскопом).
- Клинический анализ крови (лейкоцитарная формула, определение скорости оседания эритроцитов).
- Биохимический анализ крови.
- Лабораторная диагностика (проводится в целях дифференциальной диагностики) кишечных инфекций, целиакии, болезни Крона, панкреатита, пищевой аллергии.
- Определение содержания водорода, метана или меченного 14С углекислого газа в выдыхаемом воздухе.
Инструментальные методы исследования:
Еюноперфузия с использованием лактозо-солевого раствора.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Профилактика Фенилкетонурии (ФКУ) у детей:
Чтобы организовать раннюю диетотерапию и избежать тяжелых церебральных повреждений, нужно проводить массовые скрининги на фенилкетонурию в неонатальном периоде. Это позволяет также избежать нарушения функционирования печени ребенка. Чтобы оценить риск рождения ребенка с рассматриваемым диагнозом, нужно предварительное генетическое консультирование для пар, у которых уже есть ребенок с фенилкетонурией (ФКУ) или у которых есть родственники с такой болезнью.
Женщины с фенилкетонурией до момента зачатия должны строго придерживаться диеты и продолжать ее, пока будут беременными. Это позволит избежать нарушений развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Дети с ФКУ должны наблюдаться участковым педиатром и психоневрологом.
Когда следует обратиться к генетику?
Во избежание серьезных проблем о здоровье ребенка необходимо позаботиться задолго до его зачатия. Планируя беременность, осмотр и консультация профессионала не помешают, если в семье были случаи появления на свет детей с хромосомными или генетическими болезнями, а также врожденными дефектами развития. Незаменим визит к врачу при возрасте жены меньше 18 или больше 35 лет, для мужа критической планкой считается возраст после 45 лет. Показателем для посещения генетика является родственная связь между супругами, умственная отсталость жены, прием медикаментов, алкоголя и наркотиков перед зачатием, наличие вредных условий труда, тяжелая болезнь хотя бы одного из супругов в этот период и прохождение рентгеновского исследования.
В течение беременности не стоит откладывать визит, если у женщины диагностировано венерическое заболевание или вирусная инфекция, а также отмечены случаи мертворождения в прошлом. В группе риска находятся беременные, которые прошли рентгеновское обследование, принимали медикаменты без медицинской консультации или контактировали с больным краснухой.
Врач поможет выявить возможные патологии, если один из супругов страдает задержкой полового, речевого, физического или умственного развития. При этом наличие пороков может быть как единичным, так и множественным
Особое внимание уделяется пациентам с нарушениями слуха и зрения, ожирением различной степени, преждевременным половым созреванием в анамнезе и бесплодием сроком свыше двух лет. К негативным факторам относится полное отсутствие менструаций или нарушенный цикл, нестандартное строение тела, наличие хронических заболеваний, не поддающихся лечению, и самопроизвольные выкидыши
Врожденными заболеваниями занимаются врачи соответствующего профиля, у которых ребенок в обязательном порядке наблюдается после рождения.
Мифы о группах
Данная классификация приковывает уже более века людей, верящих в гороскопы и прочие закономерности. Сейчас очень распространены различные мифы. Согласно им, прописаны диеты для представителей с определенными отличиями. Якобы, они помогут сбросить лишний вес и оздоровить организм. Еще люди считают, что обладатели определенных типов имеют схожие черты характера и состояние здоровья. Можно выявить склонность к некоторым болезням по принадлежности к тому или иному типу.
Конечно, такое мнение имеет право на существование. Даже прослеживаются какие-то закономерности, доказанные научно. Но все это на веру, безусловно, приниматься это не должно. Черты характера, склонности к болезням и привычки в питании — все это очень индивидуально.

Питание и диеты
Рекомендации по составлению рациона могут различаться для людей с разными типами крови. Считается, что люди, обладающие первой группой крови резус-отрицательной склонны к быстрому набору лишнего веса. Поэтому им особенно тщательно надо следить за питанием и придерживаться здорового образа жизни. Считается, что они предрасположены ко многим заболеваниям и у них слабый иммунитет.
Особая склонность отмечается к развитию следующих заболеваний:
- сердечно-сосудистые патологии;
- болезни печени;
- сахарный диабет;
- мочекаменная болезнь;
- развитие аллергических реакций;
- инфекции дыхательных путей;
- онкологические процессы.
Людям с типом I (0) надо отказаться от любых вредных привычек (употребление алкоголя и курение). Не следует злоупотреблять высококалорийной пищей.
Лучше отказаться совсем от данной категории продуктов:
- кондитерских изделий и выпечки;
- жирной и жареной еды;
- продуктов с повышенным присутствием глютена;
- жирных молочных видов продуктов;
- кислых фруктов;
- ограничить блюда с повышенной концентрацией соли.
Следует с осторожностью употреблять продукты, вызывающие повышенное брожение в кишечнике (капуста, яблоки, кукуруза, бобы и чечевица). Считается что, люди, имеющие I (0), происходят от древних охотников
Считается что, люди, имеющие I (0), происходят от древних охотников.
В их рационе должны быть обязательно:
- нежирное красное мясо;
- субпродукты (почки, печень и другие);
- жирные виды морской рыбы (сельдь, лососевые виды, сардины);
- морепродукты (моллюски, креветки);
- листовые овощи;
- брокколи;
- сухофрукты;
- орехи;
- оливковое масло.
Для людей с данной принадлежностью нежелательно употреблять вместе мясные и молочные продукты.

Зависимость характера человека
Принято выделять некоторые особенности характера, типичные для носителей I (0). Некоторые теории приписывают лицам с 1 отрицательной группой крови определенные устойчивые характеристики. Им присущи высокое стремление непременно добиться своей цели и сильные лидерские качества.
А также для представителей с этим типом характерно:
- повышенная эмоциональность;
- повышенное стремление к самосохранению;
- сильные волевые качества;
- прогнозирование результатов своих поступков;
- склонность к ревности;
- отсутствие страха;
- доведение начатого дела до логического конца;
- негативная реакция на критические замечания;
- отсутствие возможности адаптироваться к изменчивым обстоятельствам;
- низкий уровень выносливости.
Среди носителей первой отрицательного типа попадается немало представителей, занимающих управленческие должности, спортсменов, военнослужащих, эффективных предпринимателей и политических функционеров.
Носители 1 группы крови резус-положительной имеют сходные характеристики. Им свойственно стремление к управлению людьми, высокие волевые качества и стремление к комфортным условиям.
Формы олигофрении
Всего выделяется три формы олигофрении:
- Первая. Вызвана факторами, носящими наследственный характер. К ним относятся такие заболевания, как синдром Крузона, синдром Марфана, истинная микроцефалия и прочие.
- Вторая. Характеризуется тем, что развитие олигофрении вызывается поражением плода внутри утробы. Обычно причиной этого становятся различные вирусные инфекции, токсоплазмоз, врожденный сифилис, гормональные нарушения или листериозом.
- Третья. Обычно возникает в период развития плода при воздействии таких факторов, как конфликт резус-фактора. В последовом периоде оказывает влияние родовые травмы или асфиксия. И перенесение инфекций, черепно-мозговые травмы, врожденная гидроцефалия, а также слабое развитие систем головного мозга.
Существуют также истинная и ложная формы олигофрении, которые не относятся ни к одной из вышеуказанных.
Клиническая картина
Для больных фенилкетонурией характерен светлый цвет волос и голубая радужная оболочка глаз (дети всегда светлее своих родителей и здоровых братьев и сестер). В первые 2_3 месяца жизни большинство детей выглядит совершенно здоровыми. Лишь у некоторых из них отмечается вялость, сонливость, реже беспокойство. Ранним симптомом является рвота. Отмечается своеобразный запах мочи ребенка (затхлый, мышиный). К концу первого полугодия жизни у 20—50% больных детей появляются экзематозные изменения кожи, иногда значительные, у некоторых развивается склередема (см. Склередема новорожденных). В этом же возрасте начинает выявляться задержка психического развития, к-рая, нарастая, в дальнейшем приводит к тяжелому слабоумию (см.) — имбецильности или идиотии. Лишь у незначительного числа больных поражение интеллектуальной сферы выражено нерезко. Отмечается отставание в физическом развитии. Задерживается формирование двигательных навыков. У многих больных появляются судороги, которые могут постепенно исчезать даже без лечения, обнаруживается гилеррефлексия, непроизвольные движения. У большинства больных на ЭЭГ регистрируется пароксизмальная активность. У некоторых детей отмечается спазм мышц конечностей (преимущественно ног). Все отмеченные изменения характерны для больных, не получающих с рождения патогенетического лечения.
Получение аминокислот
Аминокислоты получают различными методами, некоторые из них предназначены специально для получения тех или иных аминокислот. Наиболее распространенными общими методами химического синтеза аминокислоты являются следующие.
1. Аминирование галоидопроизводных органических кислот. На галоидопроизводное (обычно бромзамещенную кислоту) действуют аммиаком, в результате чего галоид замещается на аминогруппу.
2. Получение аминокислоты из альдегидов путем обработки их аммиаком и цианистым водородом или цианидами. В результате такой обработки получается циангидрин, который далее аминируется, образуя аминонитрил; омыление последнего дает аминокислоту.
3. Конденсация альдегидов с производными глицина с последующим восстановлением и гидролизом.
Отдельные аминокислоты могут быть получены из гидролизатов белков в виде труднорастворимых солей или других производных. Например, цистин и тирозин легко осаждаются в изо электрической точке; диаминокислоты осаждают в виде солей фосфорно-вольфрамовой, пикриновой (лизин), флавиановой (аргинин) и других кислот; дикарбоновые аминокислоты осаждают в виде кальциевых или бариевых солей, глутаминовая кислота выделяется в виде аминокислот гидрохлорида в кислой среде, аспарагиновая кислота — в виде медной соли и так далее. Для препаративного выделения ряда аминокислот из гидролизатов белка применяют также методы хроматографии и электрофореза. Для промышленных целей многие аминокислоты получают методами микробиологического синтеза, выделяя их из культуральной среды определенных штаммов бактерий.